Alberi filogenetici: come crearli con un software dedicato
Gli alberi filogenetici aiutano a visualizzare le relazioni evolutive tra specie, gruppi biologici e organismi. Con un creatore di alberi filogenetici come EdrawMax puoi organizzare dati, rappresentare ramificazioni evolutive e creare diagrammi chiari e modificabili grazie a modelli pronti, simboli personalizzabili e strumenti di esportazione.


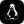



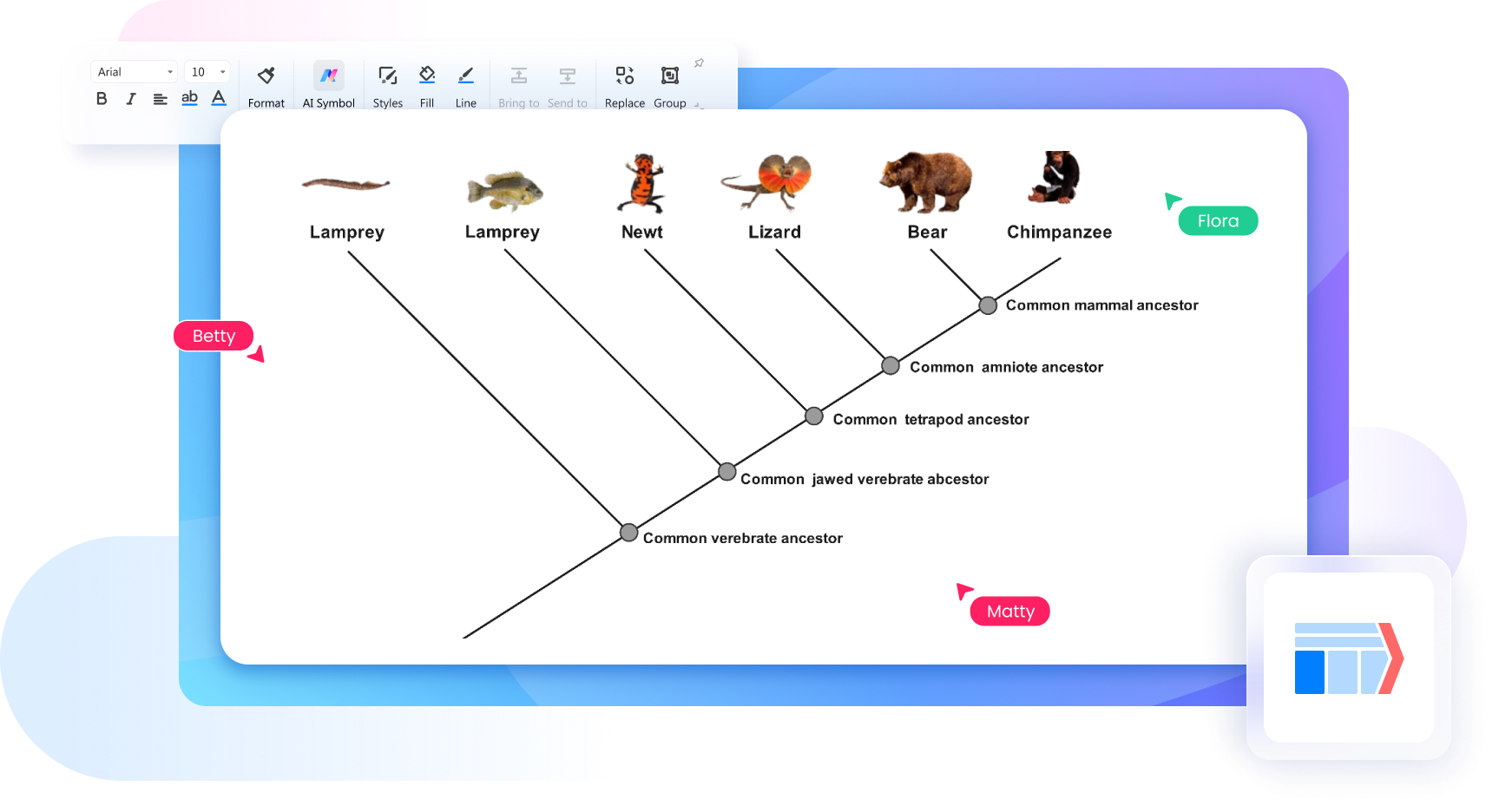
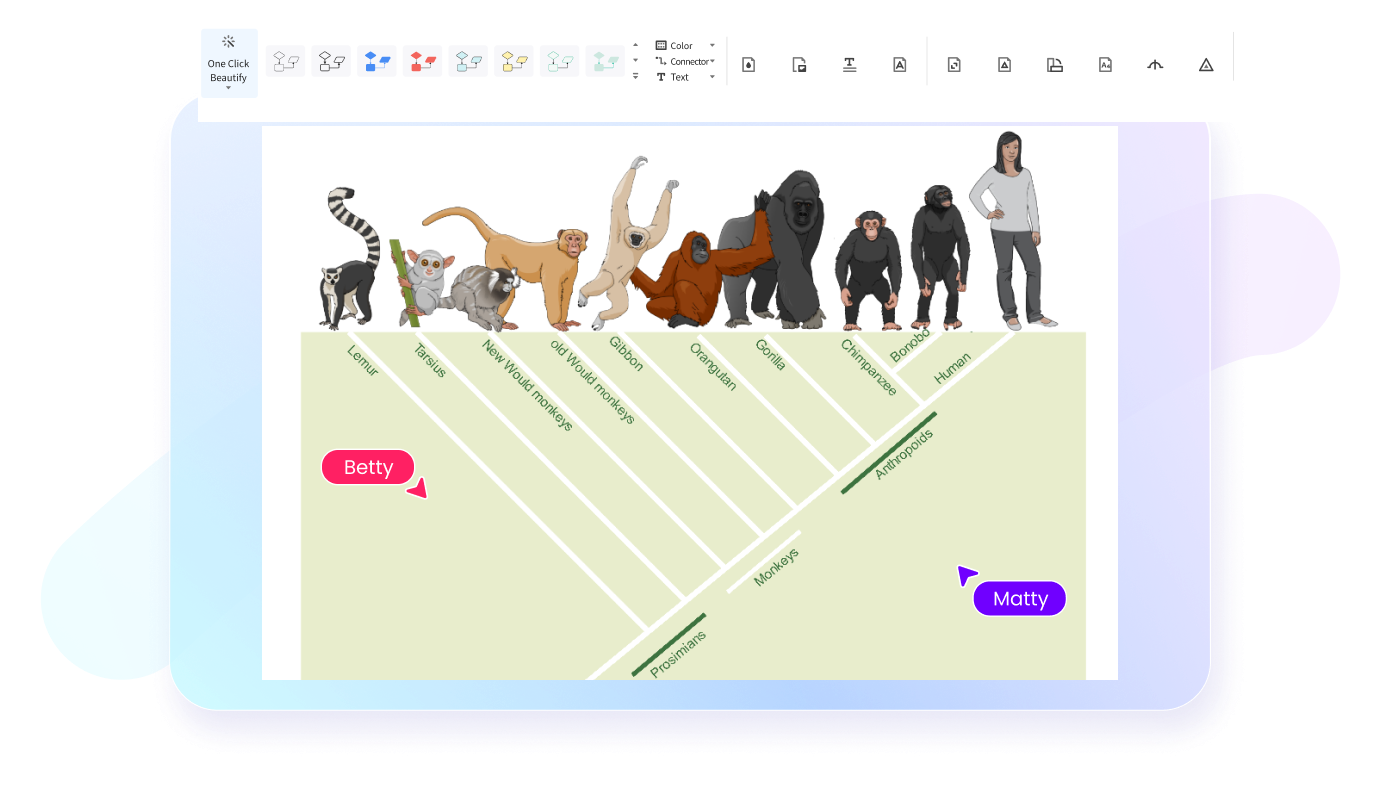
Gli alberi filogenetici sono diagrammi che mostrano le relazioni evolutive tra specie, organismi o gruppi biologici sulla base di caratteristiche condivise o dati genetici. Servono a rappresentare in modo visivo come diversi organismi possano derivare da antenati comuni e come si siano differenziati nel tempo attraverso processi evolutivi.
Un albero filogenetico può essere utilizzato nello studio della biologia evolutiva, della tassonomia, della genetica e dell’insegnamento scientifico, perché aiuta a comprendere meglio connessioni, divergenze e somiglianze tra i vari taxa. Per organizzare questi rapporti in modo più chiaro, molti utenti scelgono un creatore di alberi filogenetici o un software dedicato che permetta di disporre rami, etichette e nodi in modo ordinato e modificabile.
Gli alberi filogenetici servono a visualizzare in modo chiaro le relazioni evolutive tra organismi diversi. Sono utili per mostrare origini comuni, separazioni tra gruppi, caratteristiche condivise e possibili percorsi di evoluzione, rendendo più semplice l’analisi di informazioni biologiche complesse.
In pratica, possono essere usati nella ricerca, nella didattica, nella classificazione delle specie e nella presentazione di dati genetici o morfologici. Un diagramma per alberi filogenetici ben organizzato aiuta non solo a interpretare meglio i dati, ma anche a comunicarli con maggiore precisione in contesti accademici, scientifici o formativi.
I principali tipi di alberi filogenetici includono gli alberi radicati, gli alberi non radicati e i cladogrammi. Gli alberi radicati mostrano un antenato comune e indicano una direzione evolutiva, mentre quelli non radicati rappresentano soprattutto le relazioni tra organismi senza definire con precisione l’origine dell’evoluzione. I cladogrammi, invece, mettono in evidenza la struttura delle ramificazioni tra gruppi in base a caratteristiche condivise.
La scelta del tipo di rappresentazione dipende dall’obiettivo dell’analisi e dal tipo di dati disponibili. In alcuni casi serve evidenziare la storia evolutiva in modo dettagliato, mentre in altri è più utile mostrare in modo semplice la relazione tra specie o gruppi. Usare un software per alberi filogenetici può aiutare a scegliere un layout più adatto e a trasformare dati complessi in un diagramma leggibile e ben strutturato.
Per leggere un albero filogenetico in modo corretto, bisogna partire dai nodi, dai rami e dalle connessioni tra i diversi gruppi. I nodi rappresentano generalmente antenati comuni, mentre i rami mostrano la separazione evolutiva tra specie o gruppi biologici. Osservando il punto in cui i rami si dividono, è possibile capire quali organismi condividono relazioni evolutive più strette.
È importante ricordare che la posizione visiva dei nomi non è sempre l’elemento principale da interpretare: ciò che conta davvero è la struttura delle ramificazioni e il modo in cui i gruppi sono collegati tra loro. Un diagramma per alberi filogenetici ben organizzato aiuta a leggere con maggiore chiarezza somiglianze, divergenze e linee evolutive, soprattutto quando viene creato con un software che rende più ordinati nodi, etichette e connessioni.
Il tuo creatore di alberi filogenetici facile da usare
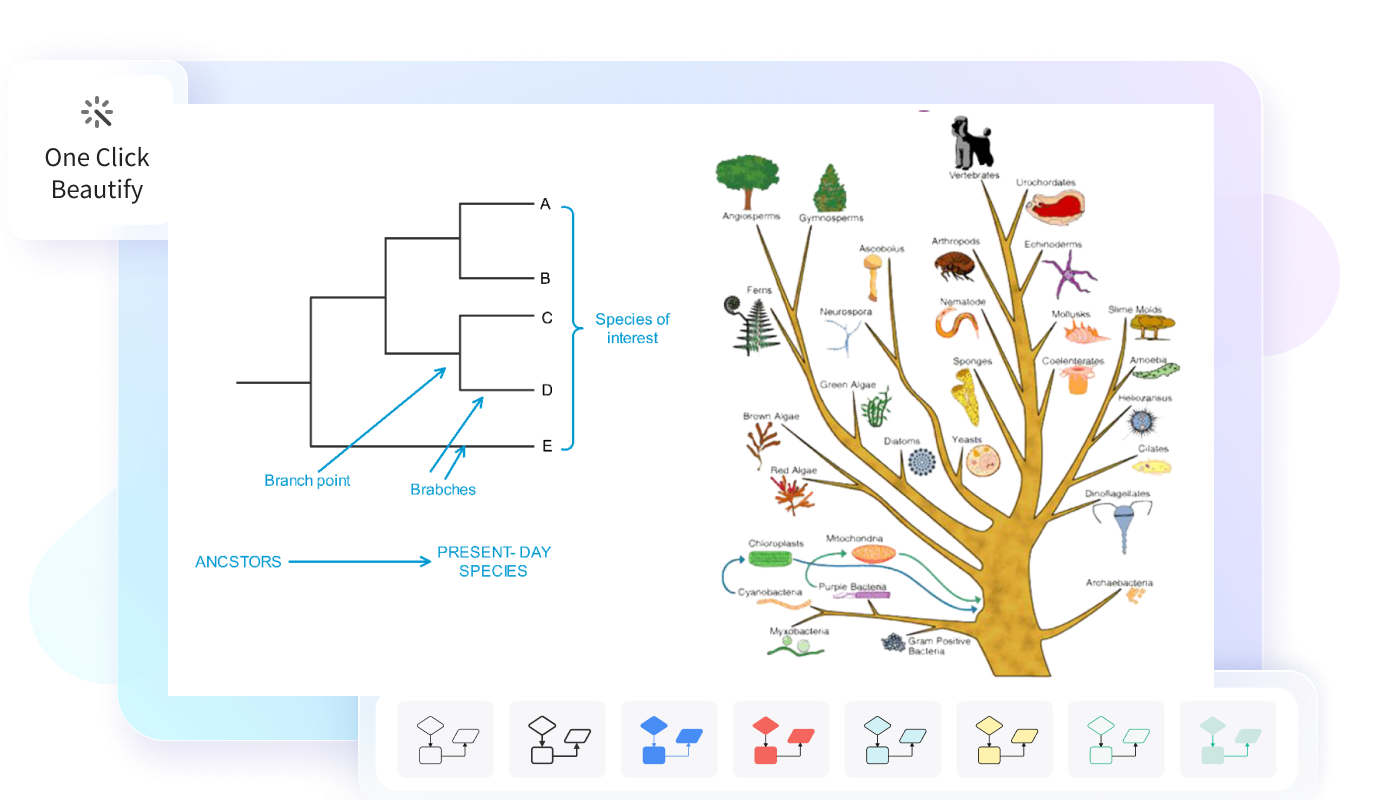
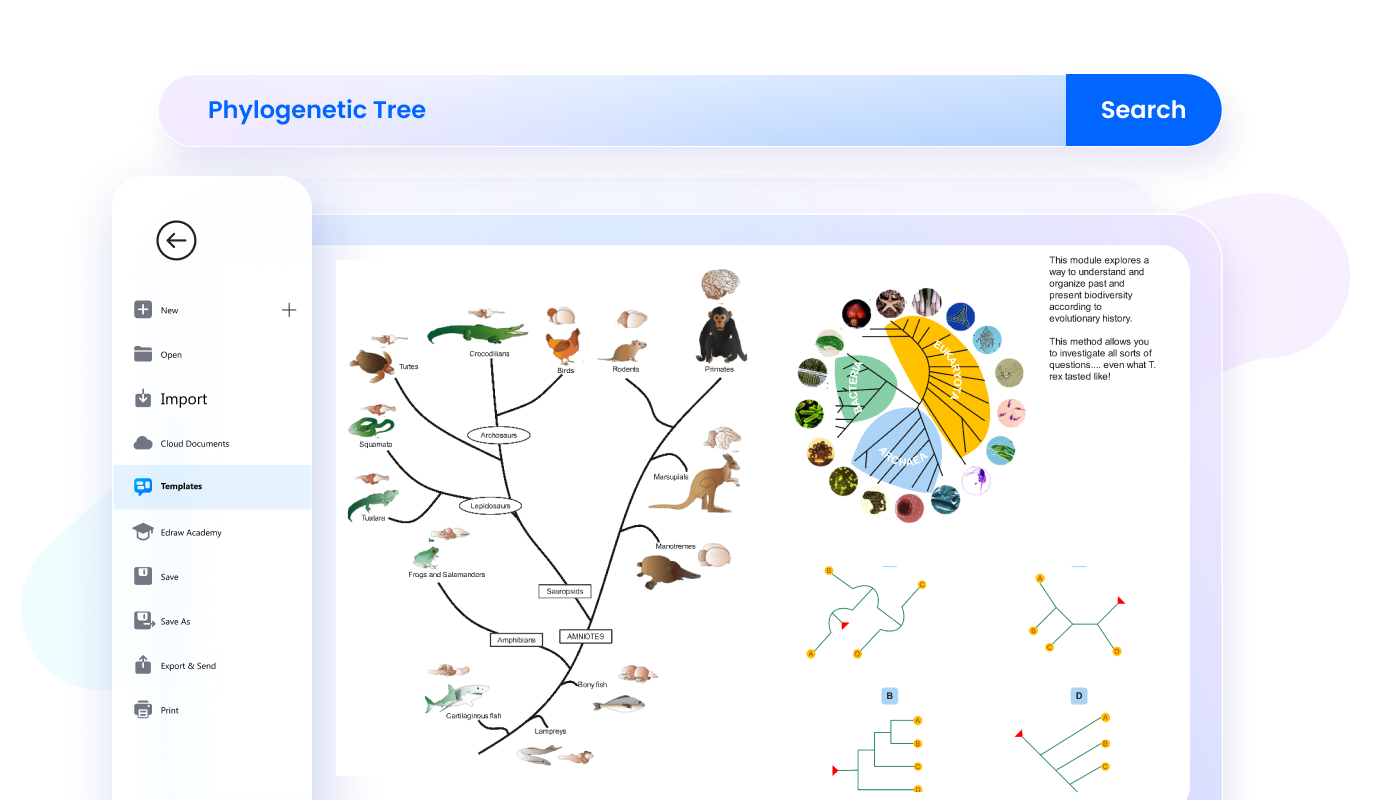
Software per alberi filogenetici per tutti



Perché i team scelgono EdrawMax
Come creare un albero filogenetico in modo chiaro?





Cosa dicono i nostri utenti












Domande frequenti sui creatori di alberi filogenetici
-
Quale software usare per creare alberi filogenetici in modo chiaro?Per creare alberi filogenetici in modo chiaro, conviene usare un software che permetta di organizzare facilmente rami, nodi, etichette e connessioni tra specie o gruppi biologici. Un creatore di alberi filogenetici con modelli pronti e strumenti di modifica aiuta a rendere il diagramma più ordinato, leggibile e semplice da aggiornare. EdrawMax è utile in questo processo perché consente di personalizzare rapidamente la struttura del diagramma e presentare i dati in modo più professionale.
-
EdrawMax offre modelli di alberi filogenetici già pronti?Sì, EdrawMax offre modelli di alberi filogenetici che possono essere modificati in base al tipo di progetto, al numero di specie coinvolte e al livello di dettaglio richiesto. Usare un modello già pronto aiuta a iniziare più velocemente e a costruire un diagramma per alberi filogenetici più coerente dal punto di vista visivo e strutturale. Questo è particolarmente utile per studio, ricerca, presentazioni o contenuti didattici.
-
Come organizzare meglio rami, nodi ed etichette in un albero filogenetico?Per organizzare meglio un albero filogenetico, è importante mantenere una struttura pulita, distribuire con ordine i rami e rendere immediatamente leggibili nodi ed etichette. Un buon software per alberi filogenetici aiuta a sistemare gli elementi in modo più chiaro, evitando sovrapposizioni e confusione visiva. In questo modo il diagramma risulta più facile da leggere, confrontare e presentare.
-
Posso collaborare con i membri del mio team su un albero filogenetico?Sì, EdrawMax dispone di funzionalità di collaborazione che consentono di condividere il diagramma, raccogliere feedback e lavorare insieme su modifiche e revisioni. Questo è utile quando l’albero filogenetico viene preparato per ricerca, presentazioni, insegnamento o lavoro di gruppo. Una collaborazione più semplice aiuta anche a controllare meglio struttura, etichette e chiarezza complessiva del diagramma.
-
Come posso condividere o esportare il mio albero filogenetico?Dopo aver creato l’albero filogenetico, puoi condividerlo con colleghi, studenti o membri del team, oppure esportarlo per revisione, pubblicazione o presentazione. Un software dedicato permette di salvare il lavoro in modo più ordinato e di esportare il diagramma in formati pratici per l’uso quotidiano. Questo rende più semplice passare dalla creazione del diagramma alla sua comunicazione o diffusione.
-
Quali metodi si usano per costruire un albero filogenetico?Tra i metodi più comuni per costruire un albero filogenetico ci sono Neighbor-Joining, Maximum Parsimony, Maximum Likelihood e Bayesian Inference. Neighbor-Joining è spesso usato per costruire alberi in modo relativamente rapido a partire da matrici di distanza, mentre Maximum Parsimony cerca la soluzione evolutiva più semplice con il minor numero di cambiamenti. Maximum Likelihood e Bayesian Inference sono approcci statistici più avanzati, utili quando si vogliono valutare con maggiore precisione i modelli evolutivi e la probabilità delle diverse relazioni tra specie.
-
Qual è la differenza tra Neighbor-Joining, Maximum Parsimony, Maximum Likelihood e Bayesian Inference?La differenza principale sta nel modo in cui ogni metodo ricostruisce le relazioni evolutive. Neighbor-Joining si basa sulle distanze tra sequenze o taxa ed è utile per ottenere rapidamente una struttura generale. Maximum Parsimony privilegia l’albero che richiede il minor numero di cambiamenti evolutivi. Maximum Likelihood valuta quale albero sia più probabile in base a un modello statistico dell’evoluzione, mentre Bayesian Inference stima la probabilità delle diverse ipotesi filogenetiche combinando dati osservati e inferenza statistica. La scelta del metodo dipende dal tipo di dati, dal livello di accuratezza richiesto e dall’obiettivo dell’analisi.